
Kandungan
Cystic fibrosis (CF) adalah penyakit yang diwarisi dan mengancam nyawa yang merosakkan paru-paru dan saluran pencernaan. Ia disebabkan oleh gen yang cacat yang memicu pengeluaran lendir menebal yang menyumbat saluran udara dan menyekat rembesan enzim pencernaan.Gejala adalah progresif dan sering parah, dan mungkin termasuk masalah pernafasan, jangkitan paru-paru berulang, pertumbuhan buruk, kemandulan lelaki, dan keradangan kronik pankreas, hati, ginjal, dan jantung.
CF boleh didiagnosis dengan ujian darah, pemeriksaan genetik, dan prosedur yang dikenali sebagai ujian klorida peluh.
Walaupun tidak ada penawar untuk CF, ada rawatan yang dapat meningkatkan panjang dan kualiti hidup seseorang.
Ini termasuk teknik pembersihan saluran udara, antibiotik yang dihirup, pengencer lendir, enzim pankreas, diet berkalori tinggi, dan ubat generasi baru yang dikenali sebagai CFTR modulator. Dalam kes yang teruk, mungkin diperlukan pemindahan paru-paru.
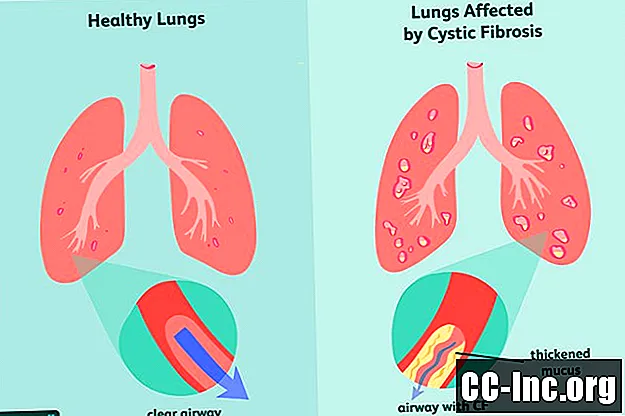
Gejala Fibrosis Kistik
Sebagai gangguan genetik, fibrosis kistik adalah sesuatu yang anda lahir. Ia mungkin atau mungkin tidak menunjukkan simptom semasa kelahiran dan selalunya boleh memakan masa berbulan-bulan atau bahkan bertahun-tahun sebelum tanda-tanda penyakit muncul. Pada masa itu, paru-paru dan saluran pencernaan mungkin sudah mengalami kerosakan yang tidak dapat diatasi.
Tanda dan gejala awal CF yang paling biasa termasuk:
- Penyumbatan najis pertama bayi (mekonium)
- Kulit rasa masin
- Batuk kronik, berdehit atau dahak berwarna
- Najis yang longgar, berminyak, dan biasanya berbau busuk
- Jangkitan paru-paru, sering berulang
- Pertumbuhan yang buruk dan kegagalan untuk berkembang maju
Kecuali gejala ini tidak dapat dikawal, tekanan pada paru-paru (dan ketidakupayaan untuk menambah berat badan) boleh memberi kesan kumulatif, mempengaruhi banyak organ dan meningkatkan risiko komplikasi penyakit.
Beberapa komplikasi yang lebih ketara termasuk:
- Akil baligh
- Bronchiectasis (penebalan kronik dinding paru-paru)
- Pengurangan berat
- Pankreatitis (keradangan pankreas)
- Kemandulan lelaki
- Hipertensi paru (tekanan darah tinggi di paru-paru)
- Batu empedu
- Diabetes yang berkaitan dengan fibrosis kistik
- Cor pulmonale (kegagalan jantung sebelah kanan)
- Sirosis (parut hati berfungsi)
Kerana CF menyebabkan kecederaan progresif pada sel dan tisu, kerosakan yang berlaku pada paru-paru dan organ lain sebahagian besarnya tidak dapat dipulihkan. Kematian selalunya disebabkan oleh kegagalan pernafasan, diikuti dengan kegagalan jantung dan kegagalan hati.
Gejala Fibrosis Kistik
Punca
Fibrosis sista disebabkan oleh mutasi gen reseptor transmembran fibrosis cyst (CFTR), yang bertanggungjawab untuk menghasilkan protein CFTR.Ini adalah protein yang diperlukan tubuh untuk mengatur aliran garam dan air masuk dan keluar dari sel . Sekiranya protein cacat atau cacat, ia boleh menyebabkan dehidrasi pada permukaan sel, menyebabkan penebalan lendir sekitarnya.
CF adalah gangguan resesif autosom, yang bermaksud bahawa anda perlu mewarisi mutasi CFTR dari ibu dan bapa anda untuk menghidap penyakit ini. Sekiranya anda hanya mewarisi satu gen yang cacat, anda tidak akan mempunyai CF tetapi akan menjadi pembawa gen yang bermutasi.
Anda boleh mewarisi penyakit ini sekiranya setiap ibu bapa anda mempunyai mutasi CFTR atau CF itu sendiri. Sekiranya kedua-dua ibu bapa adalah pembawa, anda akan mempunyai:
- 25 peratus kemungkinan mengalami CF
- 50 peratus peluang menjadi pembawa
- 25 peratus kemungkinan tidak terjejas
Sebaliknya, jika salah seorang ibu bapa anda adalah pembawa dan yang lain mempunyai CF, anda mempunyai peluang 50/50 sama ada mempunyai CF atau menjadi pembawa.
Fibrosis kistik adalah salah satu penyakit genetik yang lebih biasa, yang mempengaruhi kira-kira satu dari setiap 2.500 bayi yang dilahirkan di Amerika Syarikat.
Ini paling biasa di kalangan orang Kaukasia dan Hispanik, dan berlaku lebih jarang pada orang keturunan Afrika atau Asia.
Faktor Risiko Fibrosis KistikDiagnosis
Terdapat beberapa ujian yang digunakan untuk mendiagnosis fibrosis sista. Mereka berfungsi sama ada dengan secara langsung mengesan mutasi CFTR atau secara tidak langsung mengukur perubahan biologi yang sesuai dengan penyakit ini. Kaedah diagnosis boleh berbeza-beza semasa kehamilan, ketika bayi dilahirkan, atau bila-bila masa selepas itu.
Panduan Perbincangan Doktor Fibrosis Kistik
Dapatkan panduan cetak kami untuk temujanji doktor anda yang seterusnya untuk membantu anda mengemukakan soalan yang betul.

Dari dua ujian standard yang biasa digunakan untuk mendiagnosis CF:
- Ujian klorida peluh, juga dikenali sebagai ujian peluh, mengukur jumlah klorida pada kulit. Kerana CF mengganggu pemindahan garam ke dan dari sel, akan terjadi pengumpulan garam dalam peluh.
- Ujian CFTR genetik digunakan untuk mengesan mutasi mutasi CFTR yang paling biasa. Walaupun terdapat lebih dari 2.000 mutasi CFTR yang diketahui menyebabkan fibrosis kistik, 23 yang termasuk dalam panel standard mewakili kemungkinan tersangka.
Semasa kehamilan, ujian genetik CFTR dapat digunakan untuk menguji cairan yang diperoleh melalui amniosentesis atau sel yang diekstrak melalui persampelan chorionic villus (CVS).
Pemeriksaan bayi yang baru lahir juga digunakan secara standard untuk mendiagnosis CF dan hari ini diberi mandat di semua 50 negeri dan Daerah Columbia. Apa yang diperlukan ini akan berbeza bergantung pada tempat di Amerika Syarikat anda tinggal. Sekiranya hasil pemeriksaan bayi baru lahir positif, ujian peluh akan digunakan untuk mengesahkan diagnosis.
Bagaimana Fibrosis Kistik DiagnosisRawatan
Walaupun tidak ada penawar untuk fibrosis kistik, kemajuan dalam perawatan telah memperpanjang umur mereka yang hidup dengan penyakit ini.
Tujuan rawatan CF adalah empat kali ganda: untuk mencegah jangkitan, mengekalkan fungsi paru-paru, menormalkan pencernaan, dan memperlambat perkembangan penyakit.
Antara alat terapi yang digunakan untuk menguruskan CF:
- Teknik pelepasan saluran udara (ACT) dilakukan untuk mengeluarkan dan mengeluarkan lendir terkumpul dari paru-paru. Teknik termasuk batuk huff, perkusi dada, atau ayunan dinding dada.
- Makanan tinggi lemak dan tinggi kalori digunakan untuk mengimbangi penyerapan lemak, protein, dan nutrien dalam usus.
- Makanan tambahan enzim pankreas digunakan untuk menguatkan enzim pencernaan yang tidak dapat dihasilkan pankreas disebabkan oleh peningkatan lendir.
- Antibiotik diambil setiap hari untuk mencegah jangkitan paru-paru bakteria.
- Mukolitik- ubat yang digunakan untuk menipis lendir sebelum ACT - boleh digunakan.
- Modulator CFTR adalah kelas ubat baru yang dapat memperbaiki kecacatan tertentu dalam protein CFTR dan mengembalikan fungsi pengawalseliaannya.
- Terapi oksigen boleh digunakan semasa episod akut ketika pernafasan anda mengalami gangguan teruk.
- Pemakanan enteral, juga dikenal sebagai makan tiub, dapat digunakan jika Anda tidak dapat menjaga berat badan melalui pemakanan normal.
- Pemindahan paru-paru dipertimbangkan apabila paru-paru anda tidak lagi dapat menyokong kelangsungan hidup tanpa pengudaraan mekanikal.
Mengatasi
Pada tahun 1938, ketika fibrosis kistik pertama kali diklasifikasikan sebagai penyakit, kanak-kanak jarang hidup melebihi tahun pertama kehidupan mereka. Menjelang tahun 1980-an, seseorang boleh mengharapkan hidup selama 20 hingga 25 tahun. Hari ini, gambarannya telah berubah sepenuhnya dengan orang-orang yang hidup hingga usia 40-an dan bahkan 50-an sekiranya rawatan dimulakan lebih awal dan dipatuhi.
Ini bukan untuk menunjukkan bahawa CF adalah lebih serius daripada yang pernah berlaku. Ini adalah peristiwa yang mengubah hidup, yang memerlukan ketekunan dan konsistensi untuk tidak hanya mengatasi penyakit ini tetapi menjalani taraf hidup setinggi mungkin.
Untuk tujuan ini, anda perlu menormalkan CF dalam hidup anda dengan menetapkan rutin dan amalan untuk mengelakkan naik turun yang boleh menyebabkan tekanan dan meningkatkan kecacatan. Di antara pertimbangan tersebut, anda perlu:
- Uruskan pemakanan anda. Orang dengan CF sering memerlukan kalori dua kali sehari yang dilakukan oleh orang lain.
- Bersenam secara berkala. Rutin kecergasan idealnya melibatkan aktiviti aerobik minimum 20 hingga 30 minit tiga kali seminggu. Cari sesuatu yang menggembirakan yang boleh anda lakukan sepanjang hayat.
- Pastikan sentiasa terhidrat dengan baik. Dengan berbuat demikian, paru-paru dan usus berfungsi dengan baik. Bergantung pada usia anda, anda harus minum tidak kurang dari enam hingga lapan gelas air tinggi setiap hari.
- Lakukan pelepasan saluran udara dengan betul. Oleh kerana keperluan kesihatan anda juga berubah, mungkin juga jenis alat pelepasan yang anda perlukan. Bercakap dengan ahli pulmonologi atau ahli terapi fizikal anda jika anda tidak mencapai hasil yang sepatutnya.
- Dapatkan sokongan. Selain rakan dan keluarga, anda boleh menghubungi bab Cystic Fibrosis Foundation (CFF) yang terdekat untuk menyambung ke rangkaian sokongan di kawasan anda.
- Dapatkan bantuan kewangan. CFF menawarkan perkhidmatan yang dapat membantu keluarga mengatasi kos rawatan CF yang lebih tinggi.
Satu Perkataan Dari Sangat Baik
Walaupun pemeriksaan baru lahir telah meningkatkan kadar diagnosis CF pada bayi secara dramatik, lebih dari 25 peratus diagnosis hanya dibuat pada masa kanak-kanak, remaja, dan dewasa awal.
Ini bermasalah kerana diagnosis dan rawatan awal dapat mencegah banyak komplikasi CF yang lebih teruk sebelum kerosakan serius dapat dilakukan. Walaupun rawatan tidak dapat menghentikan atau membalikkan penyakit, ia dapat memastikan lebih banyak tahun bebas penyakit.
Untuk tujuan ini, penting untuk mengetahui gejala awal CF dan berbincang dengan doktor anda sekiranya anda mengesyaki anak anda mungkin menghidap penyakit tersebut. Ini benar terutama di negara-negara yang hanya menjalani ujian darah IRT, yang boleh mengakibatkan sebanyak 5 peratus kanak-kanak mengalami diagnosis yang tertunda atau hasil negatif-palsu, menurut penyelidikan dari Sekolah Perubatan dan Kesihatan Awam Universiti Wisconsin .
Gejala apa yang boleh anda jangkakan dengan Fibrosis kistik?